
Next: Protein folding Up: 7.5 Folding Problems in Previous: Simplifying knots
A sampling-based motion planning approach to pharmaceutical drug design is taken in [601]. The development of a drug is a long, incremental process, typically requiring years of research and experimentation. The goal is to find a relatively small molecule called a ligand, typically comprising a few dozen atoms, that docks with a receptor cavity in a specific protein [615]; Figure 1.14 (Section 1.2) illustrated this. Examples of drug molecules were also given in Figure 1.14. Protein-ligand docking can stimulate or inhibit some biological activity, ultimately leading to the desired pharmacological effect. The problem of finding suitable ligands is complicated due to both energy considerations and the flexibility of the ligand. In addition to satisfying structural considerations, factors such as synthetic accessibility, drug pharmacology and toxicology greatly complicate and lengthen the search for the most effective drug molecules.
 |
One popular model used by chemists in the context of drug design is a
pharmacophore, which serves as a template for the desired ligand
[229,339,383,860].
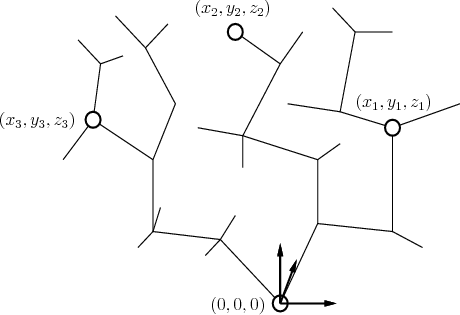
The pharmacophore is expressed as a set of features that an
effective ligand should possess and a set of spatial constraints
among the features. Examples of features are specific atoms, centers
of benzene rings, positive or negative charges, hydrophobic or
hydrophilic centers, and hydrogen bond donors or acceptors. Features
generally require that parts of the molecule must remain fixed in
![]() , which induces kinematic closure constraints. These features
are developed by chemists to encapsulate the assumption that ligand
binding is due primarily to the interaction of some features of the
ligand to ``complementary" features of the receptor. The interacting
features are included in the pharmacophore, which is a template for
screening candidate drugs, and the rest of the ligand atoms merely
provide a scaffold for holding the pharmacophore features in their
spatial positions. Figure 7.32 illustrates the
pharmacophore concept.
, which induces kinematic closure constraints. These features
are developed by chemists to encapsulate the assumption that ligand
binding is due primarily to the interaction of some features of the
ligand to ``complementary" features of the receptor. The interacting
features are included in the pharmacophore, which is a template for
screening candidate drugs, and the rest of the ligand atoms merely
provide a scaffold for holding the pharmacophore features in their
spatial positions. Figure 7.32 illustrates the
pharmacophore concept.
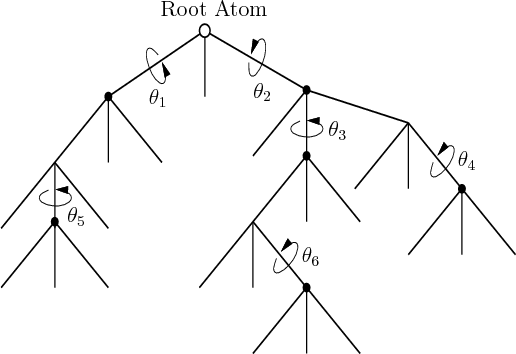
Candidate drug molecules (ligands), such as the ones shown in Figure
1.14, can be modeled as a tree of bodies as shown in
Figure 7.33. Some bonds can rotate, yielding revolute
joints in the model; other bonds must remain fixed. The drug design
problem amounts to searching the space of configurations (called
conformations) to try to find a low-energy configuration that
also places certain atoms in specified locations in
![]() . This
additional constraint arises from the pharmacophore and causes the
planning to occur on
. This
additional constraint arises from the pharmacophore and causes the
planning to occur on
![]() from Section 7.4 because the
pharmacophores can be expressed as closure constraints.
from Section 7.4 because the
pharmacophores can be expressed as closure constraints.
An energy function serves a purpose similar to that of a collision detector. The evaluation of a ligand for drug design requires determining whether it can achieve low-energy conformations that satisfy the pharmacophore constraints. Thus, the task is different from standard motion planning in that there is no predetermined goal configuration. One of the greatest difficulties is that the energy functions are extremely complicated, nonlinear, and empirical. Here is typical example (used in [601]):
Steven M LaValle 2012-04-20
![$\displaystyle \begin{tabular}{ll} $e(q)$=& $ \sum_{bonds}{{1 \over 2} K_b (R- R...
... \right] + { {c_i c_j} \over {\epsilon r_{ij} }}} \right\}. $ \end{tabular}$](img2826.gif)